עמילואידוזיס
עמילואידוזיס (Amyloidosis) היא קבוצה של מחלות המאופיינות בשקיעת סיבוני חלבונים עמילואידים ברקמות או איברי הגוף. העמילואיד נוצר עקב תקלה בקיפול סיבוני החלבון, שהמבנה השניוני שלהם נושא צורה דמוית משטחי בטא מקבילים מנוגדים. תסמיני המחלה משתנים לפי סוג המשקע והאיברים המעורבים, וכך גם הטיפול.
 | |
| מיקרוגרף המראה משקע עמילואידי ברקמת שריר הלב (צביעת קונגו אדומה) | |
| תחום |
אנדוקרינולוגיה |
|---|---|
| טיפול | |
| קישורים ומאגרי מידע | |
| eMedicine |
335414 |
| DiseasesDB | 633 |
| MeSH | D000686 |
| סיווגים | |
| ICD-10 | E85 |
| ICD-11 |
5D00 |
סיווג
עריכהעמילואידוזיס מסווגת בעיקר לפי סוג החלבון העמילואיד השוקע ברקמות, אך גם ניתן לסווג אותה לפי היקף המעורבות (מערכתית או ממוקמת), צורת הרכישה (מולדת או נרכשת), אטיולוגיות (ראשונית מול שניונית) ולפי ההסתמנות הקלינית. שיטת הסיווג הקלינית הייתה בעיקר בשימוש עד לשנות ה-70 של המאה ה-20, מאחר שבתקופה זו נפוצה הסברה כי עמילואידוזיס נגרמת על ידי סוג אחד של עמילואיד. בחלוקה לפי סוג החלבון העמילואיד, הנומנקלטורה המקובלת היא באמצעות הקידומת A לציון עמילואיד וקיצור מקובל של החלבון השוקע.
| סיווג מחלות עמילואיד לפי החלבון העמילואיד | ||
|---|---|---|
| קיצור | החלבון העמילואיד | מידע נוסף |
| עמילואדוזיס מערכתית | ||
| AL | השרשרת הקלה של אימונוגלובולין | מחלה ראשונית של הפרשת שרשראות קלות ביתר או משנית למיאלומה נפוצה |
| AA | עמילואיד הנסיוב A (SAA) | משנית למחלות דלקתיות וזיהומיות כרוניות |
| ATTR | טרנסתירטין | מחלה מולדת או נרכשת עם מעורבות לבבית ועצבית |
| Aβ2M | β2 מיקרוגלובולין | משנית לטיפול כרוני בהמודיאליזה |
| ALECT2 | גורם כמוטקטי לויקוציטי 2 | מעורבות כלייתית |
| AGel | ג'לזולין | מחלה גנטית עם ניוון של קרנית ומעורבות עצבית, עורית וכלייתית |
| AApoA1 | אפוליפופרוטאין AI | מחלה גנטית עם מעורבות כבדית וכלייתית |
| AFib | פיברינוגן A אלפא | מחלה גנטית עם מעורבות כלייתית |
| Alys | ליזוזים | מחלה גנטית עם מעורבות כבדית וכלייתית |
| עמילואדוזיס ממוקמת | ||
| Aβ | עמילואיד β | מעורבות במחלת אלצהיימר ובתסמונת דאון |
| AIAPP | עמילין | מערבות של הלבלב וקשר לסוכרת |
| APrP | פריון | מחלת קרויצפלד-יעקב וחולי מוח ספוגי מסוגים נוספים |
| ACys | ציסטטין C | אנגיופתיה עמילואידית מוחית המערבת את עורקי המוח |
| APro | פרולקטין | קשור לפרולקטינומה |
| AANF | ANP | מעורבות של העליות בלב |
| ACal | קלציטונין | קשור לסרטן מדולרי של בלוטת התריס |
אפידמיולוגיה
עריכהמעט מידע אפידמיולוגי פורסם בספרות הרפואית על עמילואידוזיס,[1] בפרט על עמילואידוזיס מערכתית, ובשל השונות הרבה בין סוגי המחלות, ישנו קושי בהתייחסות הכוללת לשכיחותן. סוג העמילואידוזיס השכיח ביותר הוא AL, עם כשני שלישים מכלל מקרי העמילואידוזיס. שיעור זה נשמר די יציב בשנות ה-2000. במקביל, שיעורי הסוג השני בשכיחותו, AA, צנחו בתקופה זו, מ-32% בראשית שנות ה-90 של המאה ה-20, לפחות מ-7% בראשית העשור השני של המאה ה-21. ייתכן שהדבר נובע מההתקדמות בטיפול במחלות הפרקים הדלקתיות והשימוש ההולך ונפוץ של תרופות ביולוגיות. לעומת זאת, שיעור האבחון של ATTR נרכש עלה מאוד, מ-0.2% לכ-6.5%, בין היתר על רקע גילוי המחלה באמצעים מתקדמים כמו דימות תהודה מגנטית של הלב.[1] הסוגים האחרים של עמילואידוזיס נדירים, ושכיחותם קטנה מ-1%.
בשתי עיירות קטנות בפורטוגל אובחנו כ-1000 חולים בפולינוירופתיה עמילואידית משפחתית, מחלה שלמעשה לא קיימת מחוץ לאזור.
אטיולוגיה
עריכהלמרות מאפיין כללי דומה של שקיעת סיבונים עמילואידים ברקמות, סוגי העמילואידוזיס השונים נבדלים בצורה ניכרת בסיבה ליצירת אותם סיבונים ולשקיעתם. עמילואידוזיס AL מתרחשת על רקע שגשוג שבט מסוים של תאי פלזמה, כמו במחלת המיאלומה הנפוצה, והפרשת שרשראות אימונוגלובולינים, לרוב שרשראות קלות, אשר שוקעות ברקמות.
עמילואידוזיס AA, הנוצרת על רקע יצירת חלבון SAA, שמופרש במצבי דלקת שונים, מופיעה במחלות דלקתיות כרוניות שונות כגון קדחת ים תיכונית משפחתית, דלקת מפרקים שיגרונית ומחלות מעי דלקתיות.
פתוגנזה
עריכהאופן היווצרות סיבי העמילואיד, כמו גם הדרך שבה הם מזיקים לתא ולרקמה, טרם הובהרו לאשורן. "השערת העמילואיד" גורסת כי חלבונים מסוימים עוברים תהליך של קיפול שגוי, אם כי הפיך, והחלבונים הללו יוצרים תלכידים אוליגומריים, ולאחר מכן פולימרים מסדר גבוה וסיבונים אשר שוקעים ברקמות. משקעים אלה שוקעים יחד עם מולקולות נוספות ומפריעים לתפקודו התקין של התא, בין היתר על ידי יצירת רדיקלים חופשיים.
משקעי הסיבונים בעמילואידוזיס מסוג AL מורכבים משרשראות קלות של אימונוגלובולינים חד-שבטיים, אשר באורכן המלא בעלות משקל מולקולרי של 23 קילדלטון. על פי רוב, מדובר בשרשראות קלות מטיפוס למבדה, אך ניתן לראות גם משקעי שרשראות קאפה, ולסוג למבדה 6 בפרט ישנו מבנה המקל על שקיעתו, בין היתר בכליות.
תסמינים קליניים
עריכההתסמינים של עמילואידוזיס כוללים:[2][3]
- עייפות קשה וחולשה
- נפיחות של קרסוליים ורגלים
- קוצר נשימה במאמץ מינימלי
- קושי בשכיבה במיטה כתוצאה מקוצר נשימה
- חוסר תחושה, עקצוץ או כאב בידיים או ברגלים, במיוחד בשורש כף היד (סינדרום התעלה הקרפלית)
- שלשולים, עם אפשרות של דימום, או עצירות
- איבוד משקל לא מכוון של למעלה מ־4.5 ק"ג
- לשון מוגדלת ונפוחה, שנראית לעיתים קרועה בשוליים
- שינויים בעור, כמו עיבוי או חבורות כתוצאה מפציעות קלות, וכתמים סגלגלים סביב העיניים.
- דופק לב לא סדיר
- קשיים בבליעה
אבחנה ובדיקות עזר
עריכהבמקרים רבים האבחנה של עמילואידוזיס מתעכבת בגלל השונות הרבה בתסמינים. אבחנה שגויה של המחלה בגלל תסמינים שמחקים מחלות פחות נדירות עלולה לעכב את מתן הטיפול המתאים. חולים הסובלים מאי ספיקת לב או אי ספיקה כלייתית מטופלים לעיתים קרובות לפני שזוהה אצלם שורש הבעיה בדרך כלל באמצעות ביופסיה. אבחנה מוקדמת ומדויקת עשויה למנוע נזקים חמורים נוספים לאיברים ולקבוע את אופן הטיפול המיטבי. לשם כך נדרשות בדיקות עזר רבות.
- בדיקות מעבדה – בבדיקות דם ושתן ניתן לזהות כמותית ואיכותית חלבון בלתי תקין בעמילואידוזיס.
- ביופסיה – בדיקת דגימת רקמה מאיבר נגוע עשויה לסייע בקביעת סוג העמילואיד ששקע באיבר. בגופם של חולים החשודים כלוקים בעמילואידוזיס מסוג AL הדרך היחידה והבטוחה לאבחון היא בשאיבת דוגמת רקמה מרקמת השומן בבטן החולה לבדיקה היסטולוגית במיקרוסקופ. בדיקה זו תהיה חיובית ב 80% מהחולים בעמילואידוזיס. לחלופין, ביופסיה מהאיבר שתפקודו איננו תקין אמינה יותר, אך מורכבת ומסוכנת יותר.
- ביופסיה של מח העצם - שיטה טובה יותר לזיהוי המחלה בחולי עמילואידוזיס מסוג AL מאחר שניתן לזהות במח העצם תאי לימפוציטים או תאי פלסמה הגורמים למחלה. כל דגימות הרקמות הנלקחות מגוף החולה נצבעות בצבע קונגו אדום שמגיב עם העמילואיד וניתנות לזיהוי במיקרוסקופ[3].
- בדיקות דימות – דימות של האיברים הנגועים עשויה לקבוע את דרגת המחלה וכוללת:
- אקוקרדיוגרפיה – טכנולוגיה המשתמשת בגלי קול (אולטרסאונד) ליצירת תמונות נעות המדגימות את פעולת הלב
- בדיקת תהודה מגנטית (MRI ) – טכנולוגיה המשתמשת בגלי רדיו ושדה מגנטי חזק ליצירת תמונה של מבנה ותפקוד הלב
- הדמיה גרעינית - בהזרקת כמויות זעירות של חומר רדיואקטיבי לוריד ניתן לזהות את סוג העמילואידים ששקעו באיבר הנגוע ולדייק את הטיפול במחלה[2].
טיפול
עריכההטיפול בעמילואידוזיס מסוג AL מתמקד בשבט תאי הפלזמה המשגשגים במח העצם, בדומה לטיפול במיאלומה נפוצה. טיפול בקורטיקוסטרואידים, ובפרט דקסמתזון העדיף על פרדניזון בהקשר זה, בשילוב עם מלפלאן במינון גבוה, ולאחר מכן ביצוע השתלת מח עצם עצמית (אוטולוגית), מביא לתגובה המטולוגית מלאה ב-40% מהמטופלים בטיפול זה. התגובה מתבטאת בירידה בעומס תאי הפלזמה והיעלמות השרשראות הקלות הנוטות לשקוע. שיפור תסמיני ניכר כחצי שנה עד שנה לאחר התגובה ההמטולוגית. עם זאת, גישת טיפול אגרסיבית זו כרוכה בסיבוכים משמעותיים, לרבות תמותה, בשל הפגיעה הראשונית של המחלה באיברים השונים, ולכן מתאימה לכ-30%–40% מהאנשים עם עמילואידוזיס מסוג AL. באנשים עם פגיעה לבבית ניכרת והפרעות קצב, נשקלת האפשרות להשתלת לב, ולאחריה השלמת הפרוטוקול להשתלת מח עצם עצמית.[3]
פרוגנוזה
עריכהלקריאה נוספת
עריכה- John L. Berk, Vaishali Sanchorawala, Amyloidosis, Harrison's Principles of Internal Medicine, 20th Edition, McGraw-Hill Companies, 2018, Vol. 1, pp. 803-809
קישורים חיצוניים
עריכה- הערך "עמילואידוזיס - Amyloidosis", באתר ויקירפואה
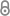 שיר־לי גולן, המחלה שגם הרופאים לא מכירים, באתר "ידיעות אחרונות", 27 באוקטובר 2018
שיר־לי גולן, המחלה שגם הרופאים לא מכירים, באתר "ידיעות אחרונות", 27 באוקטובר 2018- עמילואידוזיס, באתר אנציקלופדיה בריטניקה (באנגלית)
הערות שוליים
עריכה- ^ 1 2 Ashutosh D Wechalekar, Julian D Gillmore, Philip N Hawkins, Systemic amyloidosis, The Lancet 387, 2016-6, עמ' 2641–2654 doi: 10.1016/S0140-6736(15)01274-X
- ^ 1 2 Amyloidosis - Diagnosis and treatment - Mayo Clinic, www.mayoclinic.org (באנגלית)
- ^ 1 2 3 Kelli Miller, Amyloidosis: Symptoms, Treatments, Prognosis, Causes, WebMD (באנגלית)
הבהרה: המידע בוויקיפדיה נועד להעשרה בלבד ואינו מהווה ייעוץ רפואי.